Weyl-Kac character formula
Weyl–Kac formula, Kac–Weyl character formula, Kac–Weyl formula, Weyl–Kac–Borcherds character formula
A formula describing the character of an irreducible highest weight module (with dominant integral highest weight) of a Kac–Moody algebra. The formula is a generalization of Weyl's classical formula for the character of an irreducible finite-dimensional representation of a semi-simple Lie algebra (cf. Character formula). The formula is very robust and has been steadily applied (with increasing technical complications) to the representations of ever wider classes of algebras, see [a3] for representations of Kac–Moody algebras and [a2] for generalized Kac–Moody (or Borcherds) algebras.
Let 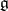 be a Borcherds (colour) superalgebra (cf. also Borcherds Lie algebra) with charge
be a Borcherds (colour) superalgebra (cf. also Borcherds Lie algebra) with charge 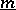 and integral Borcherds–Cartan matrix
and integral Borcherds–Cartan matrix 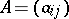 , restricted with respect to the colouring matrix
, restricted with respect to the colouring matrix 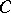 . (The charge counts the multiplicities of the simple roots.) Let
. (The charge counts the multiplicities of the simple roots.) Let  denote the Cartan subalgebra of
denote the Cartan subalgebra of 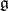 and let
and let 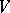 be a weight
be a weight 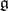 -module with all weight spaces finite-dimensional. The formal character of
-module with all weight spaces finite-dimensional. The formal character of 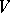 is
is
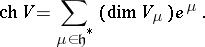 |
For 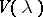 an irreducible highest weight module with dominant integral highest weight
an irreducible highest weight module with dominant integral highest weight 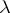 , U. Ray [a6] and M. Miyamoto [a5] have established the following generalization of the Weyl–Kac–Borcherds character formula.
, U. Ray [a6] and M. Miyamoto [a5] have established the following generalization of the Weyl–Kac–Borcherds character formula.
Let 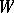 be the Weyl group,
be the Weyl group, 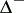 the negative roots and
the negative roots and 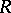 the set of simple roots counted with multiplicities. Let
the set of simple roots counted with multiplicities. Let 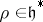 be such that
be such that
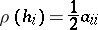 |
for all 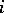 . Define
. Define 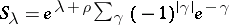 , where the sum runs over all elements of the weight lattice of the form
, where the sum runs over all elements of the weight lattice of the form 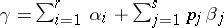 such that the
such that the 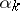 are distinct even imaginary roots in
are distinct even imaginary roots in 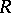 , the
, the 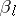 are distinct odd imaginary roots in
are distinct odd imaginary roots in 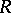 ,
,
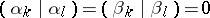 |
if 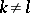 ,
,
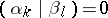 |
for all 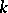 ,
,  ,
,
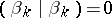 |
if 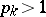 , and
, and
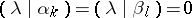 |
for all 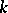 ,
, 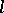 . Set
. Set 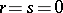 if
if 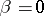 , and define
, and define 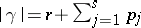 . Then
. Then
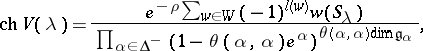 |
where 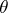 is the colouring map induced by
is the colouring map induced by 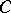 and
and 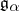 is the
is the 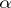 root space of
root space of 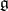 .
.
In the case of Kac–Moody algebras, there are no imaginary simple roots and 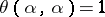 for all
for all 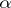 , so one recovers the Weyl–Kac formula
, so one recovers the Weyl–Kac formula
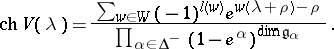 |
These character formulas may also be applied to representations of associated quantum groups where quantum deformation theorems are known (see [a4] and [a1], for example).
References
| [a1] | G. Benkart, S.-J. Kang, D.J. Melville, "Quantized enveloping algebras for Borcherds superalgebras" Trans. Amer. Math. Soc. , 350 (1998) pp. 3297–3319 |
| [a2] | R.E. Borcherds, "Generalized Kac–Moody algebras" J. Algebra , 115 (1988) pp. 501–512 |
| [a3] | V.G. Kac, "Infinite-dimensional Lie algebras and Dedekind's 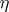 function" Funct. Anal. Appl. , 8 (1974) pp. 68–70 function" Funct. Anal. Appl. , 8 (1974) pp. 68–70 |
| [a4] | S.-J. Kang, "Quantum deformations of generalized Kac–Moody algebras and their modules" J. Algebra , 175 (1995) pp. 1041–1066 |
| [a5] | M. Miyamoto, "A generalization of Borcherds algebras and denominator formula" J. Algebra , 180 (1996) pp. 631–651 |
| [a6] | U. Ray, "A character formula for generalized Kac–Moody superalgebras" J. Algebra , 177 (1995) pp. 154–163 |
Weyl-Kac character formula. Encyclopedia of Mathematics. URL: http://encyclopediaofmath.org/index.php?title=Weyl-Kac_character_formula&oldid=11788